
何圣贵课题组在机器学习团簇化学研究方面取得新进展
低碳烷烃是工业制备高附加值化学品的重要原料,由于低碳烷烃的C−H键非常惰性,通过设计合理的催化剂实现其绿色高效转化一直是化学领域的挑战性课题,在电子结构层面理解C−H键活化的机制对于理性设计温和条件下转化烷烃的催化剂具有重要意义。催化剂表面化学键的生成和断裂一般发生在少数原子形成的活性中心处,单原子精准的金属团簇具有确定的结构和性质,是研究金属中心活化C−H键微观机制的重要模型。然而,已有的金属团簇反应研究局限于组成和尺寸缺乏关联的团簇个例,人们一般只能定性理解团簇反应性随组成和尺寸的变化,难以建立定量描述并预测金属团簇反应性的模型。
在国家自然科学基金委的支持下,化学研究所分子动态与稳态结构实验室何圣贵课题组基于自主研制的团簇仪器平台,系统制备了贵金属铑分别与前过渡金属钒和后过渡金属钴形成的成分相互关联的异核金属团簇体系,成功测定了百余例团簇与低碳烷烃反应的通道和速率,测得的速率常数跨度达六个数量级。在实验数据基础上,采用自主发展的结合迁移学习的深度神经网络方法(J. Chem. Theory Comput. 2023, 19, 1922-1930),高效搜索团簇的几何和电子结构。通过特征工程,发现了四个影响铑基金属团簇C−H活化反应性的关键电子结构特征,成功建立了定量描述随团簇尺寸变化“杂乱无章”的速率常数的机器学习模型,测试表明模型具有良好的预测能力。目前化学领域机器学习建模主要针对理论计算产生的数据,而该工作的重要特色是针对实验数据进行机器学习建模,这种研究方法有望拓展到其它团簇和分子反应体系。
相关研究成果发表在J.Am. Chem. Soc.2024,146, 12485-12495。文章的共同第一作者为博士生赵西官和杨祁,通讯作者为赵艳霞副研究员和何圣贵研究员。
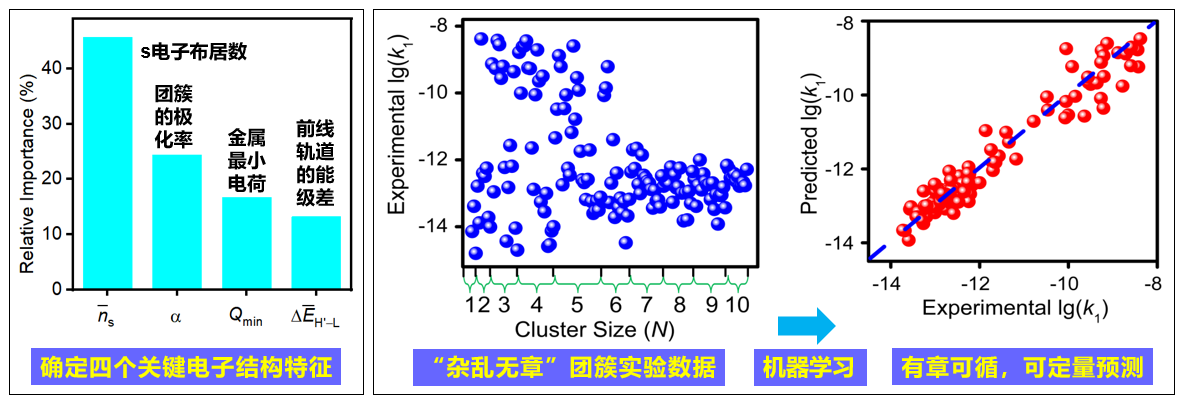
图1 基于电子结构特征(左)针对实验测定的金属团簇反应速率常数进行机器学习建模(右)

图2 金属团簇反应活性实验数据半自动化测量装置
分子动态与稳态结构实验室
2024年5月21日
附件下载:


